Morbus Fabry
M. Fabry ist eine X-chromosomal vererbte lysosymale Speichererkrankung, welche mit einem Mangel oder einer Funktionsstörung des Enzyms α-Galaktosidase einhergeht. Verursacht wird dieser Mangel durch Mutationen im Dieses Enzym ist für den Abbau von Lipiden im Körper zuständig, so dass ein Mangel oder ein Defekt zu der Akkumulation bestimmter Lipide (Glykosphingolipide) in Lysosomen und Beeinträchtigung der Zellfunktionen führen kann. Da fast alle Zellen des Körpers über Lysosomen verfügen, leiden Patient:innen mit M. Fabry häufig an einer Vielzahl unterschiedlicher Symptome und einer Multiorganerkrankung. Besonders häufig sind die Nieren, das Herz, das periphere Nervensystem, glatte Muskelzellen und Blutgefäßen betroffen, wobei die Ausprägung der Symptome auch von Aktivität der α-Galaktosidase abhängt.

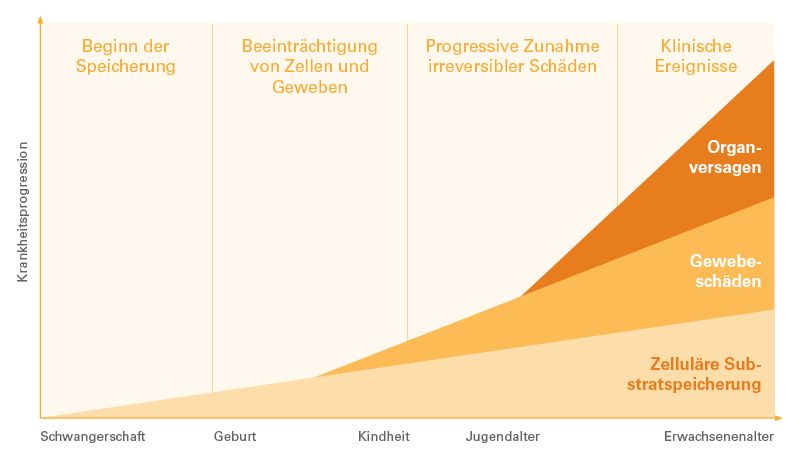
Typische Symptome in Kindheit und Jugend sind neuropathische Schmerzen (häufig Brennschmerzen an Händen und Füßen) mit Verstärkung bei Fieber, Hitze/ Kälte oder körperlicher Aktivität, eine Hypohidrose, gastrointestinale Symptome wie Abdominalschmerzen mit Durchfall, Übelkeit oder Erbrechen. Angiokeratome gehören ebenfalls zu ersten möglichen Symptomen und können sich als „Blutwarzen“ darstellen, oder als kleine rötlich-violette Flecken auf der Haut, welche vor allem zwischen dem Bauchnaben und Knien in Clustern auftreten. Fabry-typisch sind zudem wirbelartige und strahlenförmige Ablagerungen im Hornhautepithel, sogenannte Cornea verticillata.
Im Erwachsenenalter entwickeln sich typischerweise schmerzhafte (Small-Fiber-) Polyneuropathien und es kann zu einer Zunahme der Angiokeratome, Lymphödemen, einer fortschreitenden Niereninsuffizienz, Kardiomyopathien und Schlaganfällen kommen. Letztere treten meistens zwischen dem 30. Und 50. Lebensjahr auf.
Die „klassische Symptomtrias“ besteht aus Angiokeratomen, neuropathischen Schmerzen und Cornea verticillata.
Therapie des Morbus Fabry
Sobald die Diagnose eines M. Fabry anhand der typischen klinischen Präsentation und dem Nachweis einer verminderten α-Galaktosidase-Aktivität oder mittels molekulargenetischer Testung gestellt wurde, sollte in einem hierfür spezialisierten Zentrum die Einleitung einer Therapie erfolgen.
Besonders bei dieser Erkrankung ist, dass eine kausale Behandlung mittels Enzymersatztherapie (EET) zur Verfügung steht, welche die Symptome lindern und zu einem Erhalt der Organfunktionen führen kann. Zur Verfügung stehen hier zwei Präparate, welche im 2-wöchigen Abstand intravenös appliziert werden: Agalsidase α (Replagal®) und Agalsidase β (Fabrazyme®). Für bestimmte Mutationen gibt es weiterhin die Therapie mit Chaperonen (Galafold ®).
Darüber hinaus sollte eine symptomorientierte Begleittherapie erfolgen, beispielsweise mit ACE-Hemmern, β-Blockern oder AT1- Antagonisten oder bei neuropathischen Schmerzen mit Gabapentinoiden, Antidepressiva oder Opioiden.